









Edukiak
Treacher-Collins sindromea
Teacher-Collins sindromea gaixotasun genetiko arraroa da, enbrioi-bizitzan zehar garezurraren eta aurpegiaren jaiotzetiko akatsak sortzen dira, eta ondorioz aurpegiaren, belarrien eta begien deformazioak sortzen dira. Ondorio estetikoak eta funtzionalak gutxi-asko larriak dira eta zenbait kasutan esku-hartze kirurgiko ugari behar dira. Hala ere, kasu gehienetan, ardura hartzeak bizitza kalitate jakin bat kontserbatzeko aukera ematen du.
Zer da Treacher-Collins sindromea?
Definizioa
Treacher-Collins sindromea (Edward Treacher Collins-en izena, 1900ean lehenengo aldiz deskribatu zuena) sortzetiko gaixotasun arraroa da, jaiotzetik gorputzaren beheko aldean malformazio gehiago edo gutxiago larriak dituela agertzen da. aurpegia, begiak eta belarriak. Erasoak aldebikoak eta simetrikoak dira.
Sindrome horri Franceschetti-Klein sindromea edo mandibulo-aurpegiko disostosia ere deitzen zaio amaierako anomalirik gabe.
Causes
Orain arte hiru genek ezagutzen dute sindrome horretan parte hartzen dutela:
- 1. kromosoman kokatuta dagoen TCOF5 genea,
- POLR1C eta POLR1D geneak, 6 eta 13 kromosometan kokatuta hurrenez hurren.
Gene hauek aurpegiko egituren enbrioi garapenean funtsezko papera betetzen duten proteinen ekoizpena zuzentzen dute. Mutazioen bidez aldatzeak hezur-egituren (batez ere beheko eta goiko masailezurretakoak eta masailetakoak) eta aurpegiko beheko ehun bigunak (muskuluak eta azala) haurdunaldiaren bigarren hilabetean garatzen ditu. Pinna, belarri-kanala eta erdiko belarriaren egiturak (osikulak eta / edo tinpanoak) ere kaltetuta daude.
Diagnostic
Haurdunaldiaren bigarren hiruhilekoaren ekografian aurpegiko malformazioak susma daitezke, batez ere belarriko malformazio esanguratsuen kasuetan. Kasu honetan, jaio aurreko diagnostikoa diziplina anitzeko talde batek ezarriko du fetuaren erresonantzia magnetikoa (MRI) abiapuntu hartuta, malformazioak zehaztasun gehiagorekin ikustea ahalbidetuz.
Gehienetan, diagnostikoa jaiotzean edo handik gutxira egindako azterketa fisikoaren bidez egiten da. Malformazioen aldakortasun handia dela eta, zentro espezializatu batean baieztatu behar da. Odol lagin batean egindako proba genetikoa agindu daiteke, tartean dauden anomalia genetikoak bilatzeko.
Forma arin batzuk oharkabean pasatzen dira edo zorionez berandu antzemango dira, adibidez, familian kasu berri bat agertu ondoren.
Diagnostikoa egin ondoren, haurrari azterketa osagarri batzuk egin behar zaizkio:
- aurpegiko irudiak (erradiografiak, CT tomografia eta MRI),
- belarriko azterketak eta entzumen probak,
- ikusmenaren ebaluazioa,
- loaren apnea bilatu (polisomnografia) ...
Kezkatutako jendea
Treacher-Collins sindromeak 50 jaioberrietatik bati eragiten diola uste da, neskak zein mutilak. Frantzian urtero 000 kasu berri inguru agertzen direla kalkulatzen da.
Arrisku faktoreak
Erreferentzia zentro batean aholkularitza genetikoa gomendatzen da transmisio genetikoaren arriskuak ebaluatzeko.
Kasuen% 60 inguru modu isolatuan agertzen da: umea da familiako lehen gaixoa. Ernalketan parte hartu zuten ugalketa-zelulen bat edo beste ("de novo" mutazioa) eragin zuen istripu genetiko baten ondoren gertatzen dira malformazioak. Mutatutako genea ondorengoei transmitituko zaie, baina ez dago arrisku berezirik anai-arrebentzat. Hala ere, egiaztatu behar da ea gurasoetako batek sindromearen forma txikiren bat pairatzen ez duen eta mutazioa ezagutzen ez duen.
Beste kasu batzuetan, gaixotasuna hereditarioa da. Gehienetan, haurdunaldi bakoitzarekin bi transmisio arriskua bikoitza izaten da, baina tartean dauden mutazioen arabera, beste transmisio moduak daude.
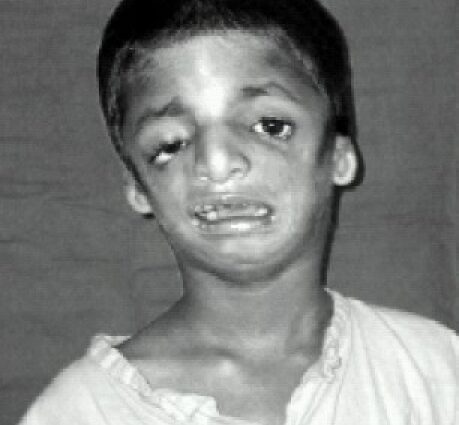
Treacher-Collins sindromearen sintomak
Kaltetuen aurpegiaren ezaugarriak maiz bereizten dira, kokots atrofiatua eta atzeratua, masail hezurrik gabeak, tenplurantz begiak beherantz okertuta, belarritakoak dituzten pabiloi txiki eta gaizki itxurako belarriak, edo erabat ez daudenak ...
Sintoma nagusiak ENT esferaren malformazioekin lotuta daude:
Arnas zailtasunak
Haur asko goiko arnasbide estuak eta ahoak estu irekita jaiotzen dira, ahozko barrunbe txikia mihiak eragozten du neurri handi batean. Horregatik, batez ere jaioberrietan eta haurtxoetan arnasteko zailtasun handiak daude, zurrungak, loaren apnea eta arnasketa ahulegia direla eta.
Jateko zailtasunak
Haurtxoetan, edoskitzea zaildu egin daiteke arnasa hartzeko zailtasunak eta ahosabaiaren eta ahosabai bigunaren anomaliak direla eta, batzuetan zatituta. Elikagai solidoak sartu ondoren elikatzea errazagoa da, baina murtxikatzea zaila izan daiteke eta hortzetako arazoak ohikoak dira.
gortasuna
Kanpoko edo erdiko belarriaren malformazioen ondorioz entzuteko ondoeza kasuen% 30-50ean agertzen da.
Ikusmenaren asaldurak
Haurren herenak estrabismoa du. Batzuk miopeak, hiperopikoak edo astigmatikoak ere izan daitezke.
Ikasteko eta komunikatzeko zailtasunak
Treacher-Collins sindromeak ez du defizit intelektuala eragiten, baina gortasunak, ikusmen arazoak, hitz egiteko zailtasunak, gaixotasunaren eragin psikologikoak eta askotan arreta mediko oso astunak eragindako asaldurak atzerapena sor dezakete. hizkuntza eta komunikatzeko zailtasunak.
Treacher-Collins sindromearen tratamenduak
Haurtxoentzako arreta
Arnasteko laguntza edo / eta hodi bidezko elikadura beharrezkoa izan daiteke haurtxoari arnasketa eta elikadura errazteko, batzuetan jaiotzatik. Arnas laguntza denboran zehar mantendu behar denean, trakeotomia bat egiten da (trakearen irekidura txikia, lepoan) kanula bat sartzeko zuzenean airea airearen pasabidea bermatuz.
Malformazioen tratamendu kirurgikoa
Ahosabai bigunarekin, masailezurrarekin, kokotsarekin, belarriekin, betazalekin eta sudurrarekin lotutako esku-hartze kirurgiko gutxi-asko konplexuak eta ugariak proposatu daitezke jatea, arnasketa edo entzumena errazteko, baina baita malformazioen eragin estetikoa murrizteko ere.
Adierazpen gisa, ahosabai bigunaren zirrikituak 6 hilabete bete baino lehen itxita daude, betazaletan eta masailetan lehenengo prozedura estetikoak 2 urtetik aurrera, mandibularen luzapena (mandibulen distrazioa) 6 edo 7 urte bitartean, berriro lotzea. belarriaren pinna 8 urte inguruan, entzumen-kanalak handitzea edo / eta osikulen kirurgia 10-12 urte inguruan ... Kirurgia estetikoko beste ebakuntza batzuk nerabezaroan egin daitezke ...
Entzungailuak
3 edo 4 hilabetetik aurrera entzumen aparatuak gorreriak bi belarrietan eragiten dituenean posible da. Kaltearen izaeraren arabera protesi mota desberdinak daude eskuragarri, eraginkortasun onarekin.
Jarraipen medikoa eta paramedikoa
Desgaitasuna mugatu eta prebenitzeko, aldizkako jarraipena diziplina anitzekoa da eta hainbat espezialistari dei egiten die:
- ORL (infekzio arrisku handia)
- Oftalmologoa (ikusmenaren asaldurak zuzentzea) eta ortoptista (begi errehabilitazioa)
- Dentista eta ortodontista
- Logopeda ...
Laguntza psikologikoa eta hezitzailea beharrezkoa izaten da.